
Родословная по генетике сахарный диабет

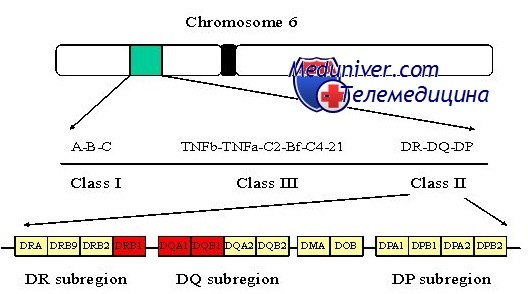
Генетика сахарного диабета I типа. Особенности наследованияСуществуют два основных типа сахарного диабета: I тип (инсулинзависимый — ИЗСД) и II тип (инсулиннезависимый — ИНСД), составляющие 10 и 88% всех случаев соответственно. Они отличаются типичным возрастом начала, конкордантностью однояйцовых близнецов и ассоциацией с конкретными аллелями главного комплекса гистосовместимости (МНС — major histocompatibility complex). Семейное накопление наблюдают при обоих типах сахарного диабета, но в одной семье обычно присутствует только I или II тип. Сахарный диабет I типа встречается в белой популяции с частотой около 1 на 500 (0,2%), в африканских и азиатских популяциях — реже. Обычно его обнаруживают в детстве или юности, и он вызван аутоиммунным поражением b-клеток поджелудочной железы, вырабатывающих инсулин. У преобладающего большинства больных детей уже в раннем детстве, задолго до развития явных проявлений болезни, вырабатываются многочисленные аутоантитела против ряда эндогенных белков, включая инсулин. Ассоциация главного комплекса гистосовместимости при сахарном диабете I типаПри I типе сахарного диабета существует подтверждение роли генетических факторов: конкордантность однояйцовых близнецов приблизительно 40%, что далеко превышает 5% конкордантности у разнояйцовых. Риск диабета I типа для сибсов больного пробанда около 7%, что дает показатель наследуемости hs = 7% / 0,2% =- 35. Давно известно, что локус МНС — основной генетический фактор при сахарном диабете, так как около 95% всех пациентов с сахарным диабетом I типа (по сравнению с примерно 50% в нормальной популяции) — гетерозиготные носители аллелей HLA-DR3 или HLA-DR4 в локусе HLA класса II в МНС [HLA — человеческие лейкоцитарные антигены (human leucocyte antigens)]. Первое исследование, показавшее ассоциацию HLA-DR3 и HLA-DR4 с сахарным диабетом I типа при использовании стандартных методов проверки достоверности различия между разными аллелями HLA, проводили методом иммунологических реакций in vitro. Позже этот метод заменили прямым определением ДНК-последовательности разных аллелей. Секвенирование локуса гистосовместимости у огромного количества больных обнаружило, что «аллели» DR3 и DR4 — не просто аллели.
Как DR3, так и DR4 могут быть подразделены на десятки аллелей, располагающихся в локусе, теперь называющемся DRB1, и определяемых на уровне последовательности ДНК. Кроме того, стало ясным, что ассоциация между определенными аллелями DRB1 и сахарным диабетом I типа частично вызвана аллелем в другом локусе класса II, DQB1, располагающимся примерно в 80 килобазах от DRB1, вместе формирующих общий гаплотип (вследствие неравновесного сцепления; см. главу 10) друг с другом. DQB1 кодирует b-цепь, одну из цепей, формирующих димер белка класса II DQ. Оказывается, что присутствие аспарагиновой кислоты (Asp) в 57 позиции b-цепи DQ тесно связано с устойчивостью к сахарному диабету I типа, тогда как другие аминокислоты в этом положении (аланин, валин или серии) определяют восприимчивость. Около 90% пациентов с сахарным диабетом I типа гомозиготны по аллелям DQB1, не кодирующим аспарагиновую кислоту в 57 положении. Раз молекула DQ, и конкретно 57 позиция р-цепи критична для связи антигена и пептида и Т-клеточного ответа, похоже, что различия в присоединении антигена, определяемые конкретной аминокислотой в 57 положении р-цепи DQ, непосредственно содействуют аутоиммунному ответу, уничтожающему инсулин-продуцирующие клетки поджелудочной железы. Тем не менее также важны другие локусы и аллели в МНС, что видно из того, что некоторые пациенты с сахарным диабетом I типа имеют в данной позиции b-цепи DQ аспарагиновую кислоту. Гены, отличающиеся от локусов главного комплекса гистосовместимости класса II при сахарном диабете I типаГаплотип МНС отвечает только за часть генетического вклада в риск сахарного диабета I типа у сибсов пробанда. Семейные исследования показывают, что даже когда сибсы имеют те же гаплотипы МНС класса II, риск болезни составляет приблизительно 17%, что значительно ниже показателя конкордантности у однояйцовых близнецов, равного примерно 40%. Таким образом, в геноме должны быть другие гены, также предрасполагающие к развитию сахарного диабета I типа и различающиеся у однояйцовых близнецов и сибсов, имеющих аналогичные условия окружающей среды. Кроме МНС, предполагают изменения еще в более чем десятке локусов, увеличивающих восприимчивость к сахарному диабету I типа, но надежно подтверждены только три из них. Это вариабельность числа тандемных повторов в промоторе гена инсулина и простой нуклеотидный полиморфизм в гене иммунного регулятора CTLA4 и в гене PTPN22, кодирующем протеин-фосфатазу. Идентификация других генов восприимчивости для сахарного диабета I типа как в пределах, так и за пределами МНС — объект интенсивного исследования. В настоящее время природа факторов негенетического риска при сахарном диабете I типа в основном неизвестна. Генетические факторы сами по себе, тем не менее, не вызывают сахарный диабет I типа, поскольку показатель конкордантности у однояйцовых близнецов составляет не 100%, а только около 40%. До получения более полной картины участия генетических и негенетических факторов в развитии сахарного диабета I типа консультирование по оценке риска остается эмпирическим. – Также рекомендуем “Генетика болезни Альцгеймера. Особенности наследования” Оглавление темы “Генетика заболеваний”:
|
Источник
Основные модели наследования
В настоящее время очевидно существование разных клинических типов СД. Однако в течение долгого времени как в клинических, так и в генетических исследованиях СД рассматривался как единая в нозологическом отношении патология. Это явилось одной из важнейших причин, затруднивших прогресс в понимании природы СД. За последние годы было подтверждено, что ИЗСД генетически отличается от ИНЗСД. При этом наибольшие успехи достигнуты в выяснении этиологии ИЗСД, хотя для окончательного установления характера его наследования необходимо больше информации. В исследовании генетики ИНЗСД, по мнению ряда авторов, прогресс незначителен.
Рассмотрим основные существующие теории и гипотезы наследования СД. М. Goodman и С. Chung (1975) были обследованы семьи 6559 больных СД с использованием метода комплексного сегрегационного анализа с целью найти различия между двумя моделями наследования: двухаллельной монолокусной и мультифакториальной.
При раннем начале СД наследуемость, вычисленная на основании мультифакториальной модели, была столь высока, что указывала на активность большого гена. Аналогичные Данные, как было показано выше, получены и нами.
В ряде работ приводятся данные, позволяющие авторам обосновать мнение о рецессивном типе наследования ИЗСД. Так, Н. Lestradet (1974), изучив родословные 1929 семей больных СД, пришел к заключению, что ИЗСД передается по аутосомно-рецессивному типу. Заболевание проявляется у 50% гомозигот, причем лишь у 1 из 12 СД развивается в возрасте моложе 15 лет.
Рецессивный тип наследования ИЗСД подтверждается в работе P. Rubinstein и соав. (1977), S. Hsu и соавт. (1977), В. Suarez и соавт. (1978). Первой группой авторов при изучении 31 семьи было найдено, что ген, предрасполагающий к СД, является рецессивным, так как пораженные сибсы были идентичны пробанду по обоим HLA-гаплотипам с достоверно повышенной частотой. Пенетрантность гена в гомозиготном состоянии, в соответствии с выводами авторов, составляет 50 %, так как 1/2 HLA-идентичных пробанду сибсов также была поражена ювенильным СД. J. Neel, однако, высказал мнение, что те же факты можно объяснить с позиций гипотезы аддитивного действия генов в одном или нескольких локусах.
В. Suarez и соавт. (1978) исследовали 24 большие семьи, сегрегирующие по ювенильному СД. Сегрегационный анализ подтвердил, что в этой группе семей ювенильный СД передается по менделевскому рецессивному типу с полной пенетрантностью. Аналогичные результаты получены ранее нами [Либерман И. С., 1965, 1970].
С помощью априорного метода было найдено, что в 34 семьях больных ювенильным ИЗСД, в составе которых насчитывалось более одного ребенка, следовало при аутосомно-рецессивном наследовании ожидать 43,23 больных детей, в действительности были больны 40 человек (34 пробанда и 6 их сибсов), что составляет 92,5% от ожидаемого (табл. 18), несмотря на заболевание лишь 6 из 60 сибсов пробандов (10%). Разница между ожидаемым и фактическим количеством больных оказалась недостоверна: x2 = 0,45; р = 0,5, что подтверждало аутосомнорецессивное наследование ИЗСД в изученной группе семей с проявляемостью гомозиготы, близкой к 100 %.
Сопоставление фактического и ожидаемого (на основании гипотезы о рецессивном наследовании) числа детей, больных СД
| Число детей в семьях (n) | Число семей (х) | Ожидаемое число больных в семьях с числом детей n (q1 • n)* | Общее число больных | |
| ожидаемое (q1 • n • х) | действительное | |||
| 2 | 20 | 1,143 | 22,86 | 22,0 |
| 3 | 10 | 1,297 | 12,97 | 13,0 |
| 4 | 2 | 1,463 | 2,926 | 2,0 |
| 6 | 1 | 1,825 | 1,825 | 2,0 |
| 10 | 1 | 2,649 | 2,649 | 1,0 |
| Всего (94) | 34 | — | 43,23 | 40,0 |
* q1 —ожидаемая часть больных детей в семьях с числом детей n; q1*n — число больных детей в семье с числом детей n по таблице С. Stern.
«Генетика сахарного диабета»,
Е.Ф.Давиденкова, И.С.Либерман
Анализ родословных, имеющихся в нашем распоряжении, подтверждает эту точку зрения. Выше были продемонстрированы родословные больных ИЗСД, в которых тяжелые ювенильные формы заболевания имели место у сибсов, а у ряда членов семей в последовательных поколениях обнаруживались различной степени нарушения ТГ — от сомнительной и диабетической гликемической кривой до манифестного заболевания, обычно ИНЗ (позднего) типа. Такой характер…
Исследователи, признающие аутосомнодоминантный тип наследования, приписывают его только части форм ИНЗСД. Некоторые авторы допускают промежуточный тип наследования и зависимость СД от дозы гена. Часть исследователей допускают трехаллельную модель наследования. Опубликованы модели наследования СД, включающие 2 и более генетических локуса. Эти модели являются переходными между моделями с моногенным и полигенным типами наследования. И, наконец, существует мнение…
В возрасте до 40 лет, в котором преимущественно развивается ИЗСД, не наблюдается существенных различий по частоте заболевания между мужчинами и женщинами, среди лиц в более старшем возрасте достоверно чаще болеют женщины. Известно, что различная поражаемость полов наиболее характерна для мультифакториально обусловленных заболеваний. В связи с этим нельзя не отметить, что различия в заболеваемости мужчин и…
Промежуточность — явление существующее, но не признанное достаточно в медицинской генетике. С. Stern (I960) писал: «Особенности, наследующиеся промежуточно, несомненно, встречаются у людей так же часто, как и среди других организмов, но твердо установленных случаев промежуточного наследования не так много. Отчасти это происходит… вследствие того, что гены, обусловливающие аномальные признаки у гетерозигот, при классификации относятся к…
Допустив, что ИЗСД детерминируется гомозиготным состоянием одного мутантного гена (аа), мы ориентировочно определили частоту генотипа аа в популяции Ленинграда, рассчитав частоту среди населения СД 1-го типа, к которому условно были отнесены все случаи СД, развившегося в возрасте до 30 лет. За основу были взяты данные двух последовательных анкетных опросов, охвативших 16 923 больных СД, состоящих…
Если частота гетерозиготного носительства составляет, по нашим расчетам, 3,284 %, то проявляемость гетерозиготного носительства равна 0,275:3,28 = 0,084, или 8,4%. Аналогичные расчеты провели R. Pieptea и I. Pavel (1968) на основании материалов Бухарестского диабетического центра. Данные, полученные нами, оказались сопоставимыми с результатами этих авторов (в %): – Бухарест Ленинград Нормальная гомозигота (АА) 97,45 96,69 Гетерозиготное…
Для полигенно обусловленных заболеваний характерно, что их частота у близких родственников пробандов превышает их частоту в популяции и убывает с понижением степени родства. При 1-й степени родства (родители и дети, братья и сестра) доля идентичных генов равна 1/2, при 2-й степени родства (дяди, тети и племянники) — 1/4, при 3-й степени родства (двоюродные братья и…
Представляет интерес исследование Н. Erlich и D. Stetler (1985), обнаруживших полиморфный сайт, локализованный около 3ʹ-конца HLA DRα гена. В этой области найдены аллели длиной 3,8 и 4,2 т. п. н. У больных ИЗСД в 30% случаев имелся аллель размером 4,2 т. п. н. S. Hodge и соавт. (1980) представили трехаллельную модель наследования ювенильного ИЗСД. Модель…
R. Spielman и соавт. (1979), допуская предрасположенность к ИЗСД, определяемую одним геном, тесно сцепленным с системой HLA, и ничтожно малую частоту рекомбинаций, нашли, что при принятых условиях имеющиеся данные более соответствуют гипотезе одной дозы рецессивного гена, чем двойной. Далее авторы использовали грубую оценку частоты генотипа, основанную на распространенности ювенильного СД и пенетрантности генотипа. Гипотеза одной…
Онкология самое страшное заболевание человечества, ухудшение состояния больного может наступить непредсказуемо в любое время, Вы можете застраховать себя и своих родных обратившись в клинику оснащенную по самым высоким стандартам высокотехнологичной медицинской помощи. Срочная госпитализация онкологических больных производится неотложно в сопровождении высококвалифицированных врачей онкологов, все необходимы анализы выполняются в считанные минуты, а атмосфера и уход в…
Источник
Родословная в генетике. Символы используемые в схемах родословнойМоногенные заболевания характеризуются определенным типом передачи в семьях. Для того чтобы установить тип передачи, обычно первым шагом получают информацию о семейной истории пациента и суммируют детали в форме родословной — графическом представлении родословного дерева, с использованием стандартных символов. Расширенная семья, изображенная на таких схемах, называется родом. Член родословной, через которого семья с генетическим заболеванием впервые попадает в поле зрения генетика (т.е. для выснения) — пробанд, если он или она болен. Человек, который обращает внимание врача на семью, советуясь с генетиком, называется консультирующийся; консультирующийся может быть больным или здоровым родственником пробанда. Родословная может иметь более чем одного пробанда, если ее учитывают более чем через один источник.
Братья и сестры называются сибсами, и семья сибсов формирует сибство. Родственников классифицируют по степеням: первая степень (родители, сибсы и потомство пробанда), вторая степень (дед, бабушка и внуки, дяди и тети, племянники и племянницы и полусибсы), третья степень (например, двоюродные братья), и так далее, в зависимости от числа членов родословной между двумя родственниками. Потомство двоюродных братьев — троюродные кузены и ребенок — двоюродный племянник кузенов его родителей.
Пары, которые имеют одного или более общих предков, считают кровнородственными. Если есть только один больной в семье, то он (или она) — изолированный или, если определено, что имеется новая мутация, — спорадический случай. Когда есть явное сходство фенотипа среди разных семей с одним и тем же дефектом, может быть использован хорошо установленный тип наследования в других семьях с тем же заболеванием как основа для диагностики и консультирования, даже если пациент представляет изолированный случай в семье. Таким образом, хотя множество пациентов с генетическими заболеваниями не имеют аналогично больных родственников, возможно признать заболевание генетическим.
При многих заболеваниях семейная передача зависит от способности больных к репродукции. Генетики используют термин «приспособляемость» как меру влияния болезни на способность к репродукции. Приспособляемость определяется как количество потомков больного с данным заболеванием, доживающих до репродуктивного возраста, в сравнении с подходящей контрольной группой. Приспособляемость — не мера физической или умственной нетрудоспособности. Например, при некотором нарушении больной может иметь нормальные умственные возможности и здоровье, но приспособляемость будет равной О, поскольку заболевание создает помехи нормальной репродукции. В других случаях серьезное инвалидизирующее генетическое заболевание может иметь нормальную приспособляемость, поскольку начало болезни наступает позже обычного возраста репродукции. – Вернуться в содержание раздела “генетика” на нашем сайте Оглавление темы “Хромосомные болезни”:
|
Источник
Учитывая разнообразные механизмы, влияющие на развитие и течение болезни, не удается проследить четких закономерностей передачи заболевания из поколения в поколение. Анализ родословных при мультифакториальных болезнях основан не на законах Менделя, как при моногенных признаках, а на эмпирически полученных данных. В результате многолетних наблюдений были выявлены следующие особенности, характерные для этой формы патологии.
- Вероятность проявления болезни зависит от степени родства с пораженным членом семьи, так как это определяет число общих генов (см. табл. 1).
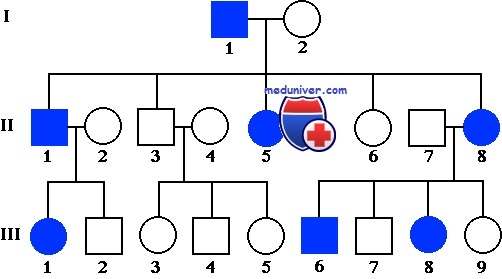
- Число больных родственников определяет прогноз для пробанда (рис. 14). Например, при сахарном диабете риск для сибсов пробанда в зависимости от числа больных родственников будет следующим: а) если родители здоровы, вероятность равна 5—10%; б) если болен один из родителей, риск равен 10—20%; в) если больны оба родителя, риск возрастает до 40%. Риск для детей пробанда в зависимости от пораженное™ его родителей составит или 10%, или 20% (рис. 15).
- Генетический прогноз зависит от степени тяжести болезни пораженного родственника, поскольку степень тяжести при мультифакториальных болезнях определяется суммарным действием нескольких генов. Так, человек, получивший 4 гена, от которых зависит артериальная гипертензия, будет иметь более тяжелую форму заболевания и, конечно, большую вероятность передачи патологического гена потомству.
- Степень наследственной отягощенности для пробанда увеличивается, если его больной родитель относится к редко поражаемому полу. Например, в случае язвенной болезни желудка и двенадцатиперстной кишки в каждой

Рис. 14. Варианты родословной с сахарным диабетом. Объяснение см. в тексте.
семье, отягощенной этим заболеванием, вероятность передачи генов потомству одинакова. Однако, вероятность заболеть всегда больше для лиц мужского пола и в том случае, если поражены женщины-родственницы пробанда. В родословной, представленной на рис. 16, риск для детей пробанда заболеть язвенной болезнью двенадцатиперстной кишки возрастает в результате двух факторов: а) оба они мужского пола (язвенная болезнь относится к заболеваниям с преимущественным поражением лишь лиц мужского пола) и б) язвенной болезнью болеет мать пробанда, т. е. в данном случае больной родитель относится к реже поражаемому полу.
Таким образом, отметим, что разнообразие типов наследственной передачи признаков в полной мере демонстрирует сложность установления типа наследования. Практический врач, не имеющий навыков в составлении и анализе родословной, не должен брать на себя смелость окончательного заключения. Ему следует направить больного и его родственников на медико-генетическое консультирование. Вместе с тем следует подчеркнуть, что для врача общей практики наиболее элементарным использованием клинико-генеалогического метода (но от этого не менее эффективным) может стать полноценный сбор данных, казалось бы, хорошо известного семейного анамнеза. Между тем и сегодня многие врачи

Рис. 15. Варианты родословной с сахарным диабетом при поражении одного или обоих родителей.
Объяснение см. в тексте.

Рис. 16. Родословная семьи с язвенной болезнью. Объяснение см. в тексте.
недооценивают значение семейного анамнеза, не имеют ясного представления, о чем нужно расспрашивать обследуемого, как оценить полученную информацию и для чего она нужна. Поэтому, если врач и собирает данные семейного анамнеза, то он часто ограничивается отдельными случайными вопросами, отмечая, например, что среди родственников были случаи заболевания пневмонией, туберкулезом, диабетом, оставляя полученные сведения без дальнейшего анализа и оценки.
Все это приобретает особое значение в настоящее время, когда перед советским здравоохранением поставлена задача обеспечить всеобщую диспансеризацию населе-
ния. При диспансеризации особенно большой интерес представляют болезни, пенетрирующие в зрелом и пожилом возрасте. В этих случаях анализ семейного анамнеза может подсказать врачу необходимость детального клинического или биохимического исследования с целью обнаружения ранних симптомов (доклинической стадии) наследственной патологии не только у конкретного индивида, но и у его родственников. Вот почему анамнез должен содержать информацию, позволяющую оценить риск заболевания для здоровых членов наследственно-отягощенной семьи. Этот риск может быть обусловлен разной степенью предрасположенности, поздней пенетрируемости или малой экспрессивностью генотипа.
Остановимся на том минимуме вопросов, которые должен задать врач обследуемому (больному или при диспансеризации).
При диспансеризации большинство обследуемых — люди, не предъявляющие каких-либо жалоб. Поэтому врач должен прежде всего, соблюдая принципы деонтологии, объяснить обследуемому, что цель вопросов заключается в выяснении предрасположенности членов его семьи к болезням тех или иных систем и органов для того, чтобы предупредить возможность их развития. Приведем примерный перечень вопросов.
Во-первых, врач должен спросить индивида, какими хроническими болезнями страдали его родственники (родители, братья, сестры, дяди, тети, племянники и др.). Если человек не может точно назвать заболевания, возможно, он знает профиль клиник (или отделений больницы), в которых лечились его родственники, или основную симптоматику болезней. Обычный ответ заключается в перечислении болезней пожилого и старческого возраста и даже в этом случае содержит некоторую полезную информацию. Однако, сбор анамнестических данных не должен ограничиваться сведениями о болезнях пожилого возраста. Не меньшее, а иногда и большее значение имеют данные о болезнях родственников в зрелом, молодом и детском возрасте, включая и врожденные физические, а также умственные аномалии. Существенными могут быть также сведения о причинах смерти родственников (инфаркт, инсульт, злокачественная опухоль, травма, туберкулез, диабет и др.) и их возрасте в момент смерти (молодой, пожилой, старый). Женщины бывают, как правило, осведомлены и о спонтанных абортах, и мертворож- дениях у их родственниц, о тех или иных пороках развития у их детей, что тоже немаловажно. Далее следует уточнить, были ли случаи выявленных в семье заболеваний единичными (спорадическими) или они повторялись у других родственников. При этом надо указать степень родства с обследуемым (отец, мать, сестра, дядя, племянник и т. д.).
Повторность заболевания в семье может быть обусловлена и неблагоприятными внешними факторами, роль которых нужно подтвердить или исключить. Так, при работе родственников на одном производстве причиной их болезни могут быть профессиональные вредности. Лекарственные воздействия на плод, так же как ряд инфекционных агентов (особенно на ранних стадиях развития), могут вызвать Пороки развития, копирующие наследственную патологию, — фенокопии. Обязательно следует получить ответы и на некоторые специфические «генетические» вопросы: о кровном родстве родителей пробанда или их происхождении из одной местности, примерном соотношении полов среди больных родственников и т. д. Это позволит с определенной вероятностью предположить тип наследования данной патологии в обследуемой семье.
Перейдем к особенностям сбора данных семейного анамнеза у больных, анализ которого в дальнейшем также должен способствовать характеристике общего генетического фона в семье, выявлению других болезней и предрасположенности к ним среди родственников. К этим особенностям прежде всего относится возможность общения с самими родственниками больного, что позволяет значительно уточнить полученную от него информацию. Это особенно важно в тех случаях, когда, руководствуясь теми или иными соображениями, больные стараются скрыть от врача известные им сведения. Вторая особенность состоит в возможности проведения обследования тех из родственников, для которых установлен высокий риск развития того или иного заболевания. Например, при выявлении нарушения толерантности к глюкозе у родственников больного сахарным диабетом целесообразно провести соответствующие профилактические мероприятия, не дожидаясь клинической манифестации заболевания.
Дать количественную оценку риска может только врач-генетик, использующий с этой целью арсенал генетических методик, включая составление родословных и определение типа наследования. Но качественно оценить риск как значительный, умеренный или малый для родственников больного или диспансеризуемого лица должен уметь каждый врач.
Объективным показателем риска иметь патологический ген может служить степень родства с больным или больными в семье. К I степени родства относятся родители, братья и сестры обследуемого. При наследственных болезнях, связанных с дефектом одного гена (моноген- ных), в этом случае вероятность иметь патологический ген составляет 1 /2 (50 %). Ко II степени родства относятся дед (бабка) обследуемого, его дяди и тетки. В этом случае вероятность иметь патологический ген составляет
(25%). При III степени родства (двоюродные братья и сестры) вероятность составляет l/g (13 %). Риск следует считать значительным при болезни родственников I и II степени родства. При наличии случаев болезни среди родственников III степени родства риск может рассматриваться как умеренный. Единичные, спорадические случаи болезни среди родственников IV и более дальних степеней родства указывают на малую степень риска.
При мультифакториальных болезнях теоретический расчет риска невозможен, поскольку и сама патология, и возможность ее проявления обусловлены сложным взаимодействием многих генов и факторов внешней среды. Однако, и здесь большое значение имеют степень родства с больными и повторные случаи патологии в семье. Например, при шизофрении, если болен один из родителей, вероятность болезни обследуемого составляет 8—12 %, а если больны оба родителя — 40 %.
Какие практические выводы может и должен сделать врач, собрав и оценив данные семейного анамнеза.
Если риск предрасположенности к семейной патологии оценивается как малый, врач может признать анамнез благополучным. Но если риск оценивается как умеренный и тем более как значительный, это свидетельствует о необходимости проведения диагностических и профилактических мер. При выявлении эти индивиды, даже если они вполне здоровы, должны быть отнесены к группе повышенного риска, взяты на учет и находиться в дальнейшем под регулярным врачебным наблюдением. Особое внимание следует обратить на профилактику, которая должна проводиться у лиц, еще не имеющих клинических проявлений болезни, но «угрожаемых» по данному заболеванию, например, при обнаружении основных «факторов риска» ишемической болезни сердца у родст
венников I степени родства пробанда с данной патологией; у лиц с высоким уровнем мочевой кислоты у членов семей, в которых есть больной подагрой, и т. д. Выявление «угрожаемых» по различным наследственным болезням лиц проводится при профилактических осмотрах населения, при популяционных исследованиях в результате тотального и селективного скрининга. Однако, использование клинико-генеалогического метода позволит решать эту задачу более эффективно. При этом целесообразно в зависимости от вида возможной наследственной патологии относить людей к определенным группам риска (по моногенным и хромосомным болезням, по заболеваниям с наследственной предрасположенностью).
Источник